општост
Тубуларна склероза је генетска болест која погађа неколико органа и ткива људског тела. Из тог разлога, он представља широк спектар симптома, од којих су неки типични за рано детињство, други за одрасле. Тубуларна склероза се може пренети са родитеља на децу, али може се јавити и због спонтане мутације ДНК.

Шта је тубуларна склероза
Тубуларна склероза је генетски поремећај који карактерише стварање хамартома у различитим органима или ткивима.
Хамартома идентификује подручје ткива у којем су се ћелије умножиле прилично интензивно, формирајући очигледну масу, сличну квржици или гомољи . Хамартоми памте туморе, али их не треба мешати: у ствари, ћелије хамартома су идентичне онима ткива у којима се размножавају; оне тумора, с друге стране, имају различите карактеристике. Међутим, треба имати на уму како те ћелије могу еволуирати и довести до бенигних неоплазми, фиброида и ангиофиброма .
Мозак, кожа, бубрези, очи, срце и плућа су највише погођени окрузи, али нису једине локације. Због мноштва укључених органа и ткива, тубуларна склероза се назива и мултисистемска генетска болест .
Касније ћете разумети зашто се хамартоми појављују само у одређеним подручјима.
епидемиологија
Учесталост и број случајева у свијету су неизвјесни. Несигурност је последица чињенице да многи пацијенти не показују симптоме и воде нормалан живот.
Међутим, процењује се да је инциденција тубуларне склерозе један на сваких 5.000-10.000 новорођенчади. У свету има око два милиона случајева.
узрок
Тубуларна склероза је генетска болест; то значи да се ген, присутан у ДНК захваћеног субјекта, променио.
Постоје два гена који узрокују тубуларну склерозу када су под утицајем њихових мутација:
- ТСЦ1 .
- ТСЦ2 .
До сада уочени случајеви тубуларне склерозе су мутирали само један од ових гена. Према томе, појединачна мутација ТСЦ1, или ТСЦ2, довољна је да се одреди тубуларна склероза.
Студије спроведене у Европи и Сједињеним Државама показују да је мутација у ТСЦ2 (80% случајева) много чешћа него у ТСЦ1 (преосталих 20%).
ТСЦ1 и ТСЦ2
ТСЦ1 ген се налази на хромозому 9 и производи протеин зван амартина .
ТСЦ2 ген се налази на хромозому 19 и производи протеин зван туберин .
Произведени протеини, амартина и туберина, уједињују се и раде заједно. Ово објашњава зашто мутација једног или другог одређује исту патологију.
ФУНКЦИЈА ТСЦ1 И ТСЦ2
Они се сматрају туморским супресорским генима и имају фундаменталну улогу у процесима:
- Раст и диференцијација ћелија током ембриогенезе.
- Синтеза протеина.
- Аутофагију.
Када се мутирају ТСЦ1 и ТСЦ2, произведени протеини су дефектни и ови физиолошки процеси се више не одвијају редовно.
| Укључени гени | ||
| ТСЦ1 | ТСЦ2 | |
| седиште | Хромозом 9 | Цхромосоме 16 |
| Произведен протеин | Амартина | туберин |
| функција | Раст и диференцијација ћелија, током ембриогенезе Синтеза протеина аутофагију | Раст и диференцијација ћелија, током ембриогенезе Синтеза протеина аутофагију |
| Проценат случајева | 20% | 80% |
ОСИГУРАЊЕ АМАРТОМА
Хамартоми могу настати када се мутација догоди у гену који контролише раст и диференцијацију ћелија, као што је ТСЦ1 или ТСЦ2. Сходно томе, ћелије расту у броју, стварајући очигледне масе; тако се формирају плакови са обликом сличним груда или гомоља У хистологији, овај процес се дефинише као хиперплазија .
ГЕНЕТИКА
Два простора:
- Сваки ген ДНК присутан је у две копије. Ове копије се зову алели .
- Људска бића имају 23 пара хромозома. Од тога, само један пар одређује пол (хромозоми пола); све остале се називају аутосомним хромозомима .
Тубуларна склероза је аутосомно доминантан генетски поремећај . Из тог разлога, довољно је да се алел промени тако да цео ген не ради како треба. У ствари, мутирани алел има више снаге од здравог ( доминација ).
У ствари, поремећаји тубуларне склерозе се погоршавају када су оба алела ТСЦ1 или ТСЦ2 мутирана. Другим ријечима, само један алел, иако доминантан с друге стране, не изазива очите симптоме. У овим случајевима говоримо о непотпуним доминантним алелима.
Наследство? ИЛИ СПОНТАНА МУТАЦИЈА?
Мутација ТСЦ1, или ТСЦ2, може настати услед:
- Наслеђена трансмисија (тј. Од једног од два родитеља) мутираног алела.
- Спонтана мутација алела у ембрионској фази (или ембриогенези).
Трећина случајева тубуларне склерозе је последица наследне трансмисије. У овим случајевима, довољно је да родитељ има мутацију ТСЦ1 или ТСЦ2 гена да би потомство било захваћено болешћу (ми смо заиста видели да тубуларна склероза је аутосомно доминантна наследна болест).
Преосталих 2/3 случајева је резултат спонтане мутације током ембрионске фазе.
| Порекло мутације | Број предмета | Мутирани ген |
| Наследна трансмисија | 1/3 | ТСЦ1 у 50% ТСЦ2 у преосталих 50% |
| Спонтана мутација | 2/3 | ТСЦ2 у 70% ТСЦ1 у 30% |
ЗАШТО СУ ОРГАНИ УКЉУЧЕНИ САМО?
Премиса: у раним фазама свог развоја, ембрион има три слоја ћелија:
- Ектодерма, највише спољашња.
- Месодерма, централна.
- Ендодерм, најдубљи.
Од сваког слоја потичу специфични органи и ткива.
| Ћелијски слој ембриона | Главни органи или ткива која потичу |
| ецтодерма | Нервни систем епидермис Епител уста Епител дебелог црева Рожница и кристал Зубна цаклина Дермалне кости |
| мезодерма | срце рене Интестинална зидна облога Мишићавост удова Серозне мембране плућа (плеура) и срца (перикард). |
| ендодерм | јетра панкреас Пробавни систем |
Ми сада поседујемо све елементе да схватимо зашто хамартоми настају само у одређеним областима тела.
ТСЦ1 или ТСЦ2 мутације се јављају у ембрионској фази ектодермних и мезодермних ћелија. Стога ће ткива, која ће бити рођена из ових ћелијских слојева, представљати хамартоме.
simptomi
Да сазнате више: тубуларна склероза - узроци и симптоми
Постоје бројни органи и ткива захваћени тубуларном склерозом. Највише погођене области су:
- Мозак, кожа, бубрези, срце, очи
Али не смијемо заборавити друге, рјеђе поремећаје:
- Плућа, црева, јетра, зуби, ендокрини систем, кости
Неки симптоми се појављују у младој доби, други у одраслој доби.
ИНЦОМПЛЕТЕ ДОМИНАНЦЕ
Већ је горе поменуто да је доминација мутираног алела ТСЦ1 или ТСЦ2 гена непотпуна. То значи да је здрав алел још увек у стању да произведе "здрав" протеин (амартина или туберина), мада у мањим количинама. Присуство "здравог" протеина надокнађује оштећење узроковано мутираним протеином. Под овим условима, хамартоми још увек не изазивају драматичне манифестације.
У тренутку када се други алел промијени (то је риједак али могући догађај), хамартоми расту неконтролирано.
ПРОФИЛАЦИЈЕ КОЖЕ
Око 90% пацијената има промене на кожи. Догађаји су бројни и разнолики. Типичне су депигментисане мрље, прингле лојне аденоме и Коененови тумори ноктију.
Депигментисана места су хипомеланотичне тачке, тј. Са мањим садржајем меланина
Прингле лојни аденоми су бенигни тумори који се називају и ангиофиброми лица . Хамартоми се појављују као мале, глобуларне масе светло црвене боје. Коененови тумори ноктију су фиброиди и потичу од хамартома од неколико милиметара.
Фотографија на кожним манифестацијама тубералне склерозе
Табела показује бројне кожне манифестације због тубералне склерозе:
| Кожна манифестација | седиште | фреквенција | Старост појављивања |
| Хипомеланотиц стаинс | стабло Уметност | 80-90% | 0-15 година |
| Прингле себацеоус аденомас (или ангиофиброма лица) | гузови насо брада | 80-90% | 3-5 година; пубертет |
| Влакна за нокте (од Коенен) | Ноге и ноге | 40-50% | > 15 година |
| Фиброус плакуе | предњи Скалп | 25% | рођење |
| Кнурлед плате | стабло Дорсо-лумбални регион | 20-40% | 2-3 године |
| Кожни фиброиди | врат ramena | заједнички | > 5 година; пубертет |
| Енамел лезије | зуби | заједнички | > 6 година |
| Муцоус фиброидс | уста | заједнички | Прве године живота |
| Орални псеудофиброми | Предња гингива усне непце | заједнички | Прве године живота |
НЕУРОЛОШКИ СИМПТОМИ
Мјеста мозга погођена тубуларном склерозом су:
- Мождана кора
- Бела супстанца
- Тхе вентрицлес
- Базални ганглији
Две бројке помажу читаоцу да разуме укључене области.

У зависности од локације и облика хамартома, могу се јавити различити поремећаји, као што су:
- епилепсија
- Субепендимал нодулес
- Тумори мозга типа астроцитома
- Ментални, бихевиорални и учење дефицити.
| епилепсија | |
| Хамартома схапе | луковица |
| Регија мозга је погођена | кора |
| фреквенција | 80-90% |
| znaci | Криза заплене:
|
| Старост појављивања | Рано дјетињство (грчеви), 75% Старост одраслих (делимично), 25% |
| Субепендимал нодули (НБ: епендима је епител вентрикула) | |
| Хамартома схапе | израслина |
| величина | <1 цм |
| Регија мозга је погођена | вентрикуле |
| фреквенција | 80-90% |
| Старост појављивања | детињство |
| komplikacije | Опструктивна хидроцефалус Еволуција у субпендималном астроцитому Цисте мозга |
| Субепендимал астроцитомас ин гиант целлс (СЕГА) \ т | |
| Хамартома схапе | израслина |
| величина | > 1 цм |
| Регија мозга је погођена | Вентрицоли (Форами ди Монро) |
| фреквенција | 6% |
| Старост појављивања | Између 4 и 10 година |
| znaci | главобоља повраћање конвулзије Измене визуелног поља Нагле промене расположења |
| komplikacije | hidrocefalus Цисте мозга |
| Ментални недостатак: | фреквенција | Тип догађаја | Старост појављивања | znaci |
| Поремећаји учења | 50% | Ментални хендикеп | Рано детињство (0-5 година) | Захтева надзор (85%) Одсуство језика (65%) Није самодовољно (60%) |
| Поремећаји понашања | 30% | аутизам Дефицит пажње хиперактивност агресивност Селф-сакаћење Поремећаји спавања | детињство | Удруживање са епилепсијом Тешко управљање породицом и школом |
КИДНЕИ ЛЕСИОНС
Врло су честе. Заправо, појављују се у 60-80% случајева. Састоји се од:
- Хамартоми слични бенигним туморима.
- Малформације реналне структуре.
| Тумор хамартома | |
| тип | Ангиомиолипом (60-70%) ангиолипома Миолипоми |
| Кратак опис | То су бенигни тумори који се појављују у вишеструком облику |
| симптоматологија | У детињству: Асимптоматско У одраслој доби: Могућа руптура хамартома, затим крварење, хематурија и бол у трбуху. |
| компликација | Поремећај бубрега |
| Малформација бубрежних структура | |
| тип | Потковасти бубрег Полицистични бубрег Недостатак бубрега (бубрежна агенеза) Доубле уретер |
| Кратак опис | Реналне цисте могу настати због тога што су ген ТСЦ2 и ген ПКД1, који одређује полицистични бубрег, један поред другог на хромозому 16. ТСЦ2 мутација такође може да утиче на ПКД1. |
| компликација | Поремећај бубрега |
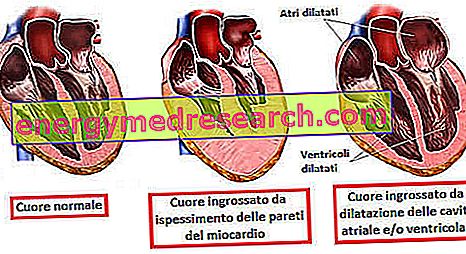
ЦАРДИОВАСЦУЛАР ЛЕСИОНС
И у овом случају, то је због хамартома сличних бенигним туморима, који се називају рабдомиоми.
| рабдомиом | |
| седиште | Зидови и шупљине срца |
| Кратак опис | Састављена је од ћелија од неколико центиметара. Регресира спонтано |
| Старост појављивања | Од рођења |
| симптоматологија | Током детињства: Симптома. Ако су димензије значајне:аритмије Поремећаји срчаног протока |
| компликација | Затајење срца |
ПУЛМОНАРИ ИЊУРИЕС
Они су углавном последица плућне лимфангиолеиомиоматоматозе ( ЛАМ ) и, у мањој мери, микронодуларне мултифокалне хиперплазије . Они су типичне манифестације одраслог доба.
| Лимпхангиолеиомиоматосис (ЛАМ) | |
| Главне карактеристике | Ретка болест Посебно погађа одрасле жене Појављују се плућне цисте Већина случајева је асимптоматска Симптоми су диспнеја попут астме, кашаљ, спонтани пнеумоторакс, респираторна инсуфицијенција |
| Микронодуларна мултифокална хиперплазија | |
| Главне карактеристике | Ретка болест Она погађа углавном одрасле, мушкарце и жене Појављују се чворићи, видљиви са рендгенским снимком груди Скоро увек асимптоматски |
ОТХЕР ИЊУРИЕС
| Локација лезије | Тип хамартома / тумора | фреквенција | догађај |
| око | Ретинал хамартома Ретинална астроцитома | 10-50% | Оштећење вида, ако хамартом или тумор утичу на макулу |
| црево | Интестинални полипи Интестиналне цисте | > 50% | без симптома |
| јетра | ангиомиолипом ангиом | <30% | без симптома |
| кости | Псеудо-цисте у рукама и стопалима | рара | без симптома |
| Ендокрини систем | аденом ангиомиолипомас | рара | без симптома |
дијагноза
Дијагноза се састоји од:
- историја
- Клиничка анализа наведених знакова
- Инструментални прегледи
ИСТОРИЈА
Лекар врши истрагу о породичној анамнези пацијента да види да ли је тубуларна склероза наследна или због спонтане мутације.
КЛИНИЧКА АНАЛИЗА ЗНАКОВА
1998. године група међународних лекара установила је дијагностички критеријум на основу горе наведених клиничких манифестација. Подељени су на:
- Главни знаци (или критеријуми)
- Мањи знаци (или критеријуми)
| Дијагноза је | |
| церта | Ако се пацијент покаже
|
| вероватан | Ако пацијент показује 1 главни и 1 мали знак |
| Могуће (сумњиво) | Ако се пацијент покаже
|
Класификација знакова је следећа:
| ГРЕАТЕР СИГНС | МИНОР СИГНС |
| Ангиофиброми лица | Вишеструке случајне повреде зубне цаклине |
| Наил или периунгуал фиброидс | Хамартоматозни ректални полипи (тј. Због хамартома) |
| Хипомеланотиц спотови (најмање 3) | Бисте цисте |
| Тектуред стаин | Радијалне линије миграције беле супстанце |
| Цортицал туберс | Гингивални фиброми |
| Субепендимал нодулес | Не-бубрежни хамартоми (или екстра-бубрежни) |
| Појединачне или вишеструке срчане рабдомиоме | Не-ретинални акромни фластери |
| Пулмонари лимпхангиолеиомиоматосис | Цонфетто хипомеланотиц скин лесионс |
| Астроцитома субгестимал гигант целл (СЕГА) | Вишеструке бубрежне цисте |
| Ренал ангиомиолипома | Породична историја |
| Вишеструки ретинални хамартоми |
ИНСТРУМЕНТАЛНИ ПРЕГЛЕДИ
| Алат за испитивање / дијагностику | Зашто се покреће? | Да ли је инвазивна? |
| офталмоскопија | Да бисте видели лезије ретине | не |
| Воодова ултраљубичаста лампа | Тражити хипомеланотичне мрље коже | не |
ЦТ мозга Нуклеарна магнетна резонанца | За претрагу:
| Да (јонизујуће зрачење) не |
| електроенцефалограм | Када пацијенти показују нападаје | не |
| Ренални ултразвук | Да бисте видели ангиомиолипоме бубрега | не |
| електрокардиограм | За откривање срчаних аритмија | не |
| ехокардиографија | Да би се откриле срчане рабдомиоме | не |
спирометрија Рендгенски снимак груди | Да бисте потражили присутност:
| не Да (јонизујуће зрачење) |
ГЕНЕТИЧКИ ТЕСТ
Ово је дуга анкета, која траје неколико мјесеци. Стога није корисна за рану дијагнозу. Радије служи за потврду дијагнозе на основу клиничких знакова.
терапија
Не постоји специфичан и ефикасан лек, јер је тубуларна склероза:
- Генетска болест.
- Мултисистем дисеасе.
Међутим, неки симптоми могу бити ограничени како би се избегле компликације и побољшао квалитет живота пацијената.
ФАРМАКОЛОШКИ ТРЕТМАН
Клиничке манифестације које се могу лечити применом лека су:
- Инфантилна епилепсија
- Плућна лимфангиолеиомиоматоза (ЛАМ)
- Поремећаји бубрега
Инфантилна епилепсија . Мали пацијент добија антиконвулзивне лекове:
- АЦТХ (адренокортикотропни хормон)
- вигабатрин
Пулмонари лимпхангиолеиомиоматосис . Корисни су бронходилататори, бета-2 агонисти као што је салбутамол. Ефикасност хормонске терапије на бази прогестерона или бусерелина је неизвесна
Поремећаји бубрега . Користе се антихипертензиви као што су АЦЕ инхибитори и диуретици.
ФИЗИКАЛНО-ХИРУРШКИ ТРЕТМАНИ
Они се састоје од интервенција чији је циљ уклањање:
- Ангиофиброми лица
- Влакна за нокте
- Кожне плоче
- Коцкасте мрље
- Субепендимал астроцитомас ин гиант целлс (СЕГА) \ т
- Ренал ангиомиолипомас
- Лезије плућа
- Гомољи церебралног кортекса, који узрокују епилепсију
Следећа табела сумира главне терапијске третмане и њихове карактеристике.
| симптом | лечење | МИЦРОдентистри |
| Ангиофиброми лица | ласер терапија | Минимално инвазивна |
| Влакна за нокте | дијатермија Криотерапија Хируршко уклањање | не Минимално инвазивна да |
| Тектуред спотс | ласер терапија Хируршко уклањање | Минимално инвазивна да |
| Кожне плоче | Криотерапија | Минимално инвазивна |
| Субепендимал астроцитомас ин гиант целлс (СЕГА) \ т | Хируршко уклањање | да |
| Ренал ангиомиолипомас | Артеријска емболизација | да |
| Плућна лимфангиолеиомиоматоза (тешка) | Трансплантација плућа | да |
| Церебрални кортекс | Хируршко уклањање | да |
Праћење и прогнозе
Премиса: медицинско праћење се односи на пацијента који је, оболел од рака, подвргнут хируршкој операцији.
Периодичне провере се препоручују за праћење . Офталмоскопија, тј. Преглед очне главе, може се вршити и једном годишње. Супротно томе, неуролошка, срчана и бубрежна стања захтевају чешће праћење.
ПРОГНОЗА
Еволуција тубуларне склерозе је променљива и зависи од сваког случаја.
Неки пацијенти показују благе, готово неприметне симптоме. За њих, квалитет живота не утиче на болест, а прогноза је одлична.
Насупрот томе, други пацијенти показују много драматичнију и очигледнију симптоматологију. Смрт долази углавном због неуролошких лезија, па прогноза постаје веома неповољна.
ГЕНЕТИЦ ЦОНСУЛТИНГ
Ако један родитељ има тубуларну склерозу, вјероватноћа да дијете наслиједи исто стање је 50%.
Ако је, с друге стране, захваћено дијете здравих родитеља, вјероватноћа да се друго дијете разболи је врло ниска. У овим случајевима, генетски тест разјашњава да ли су родитељи носиоци тубуларне склерозе, или је дошло до спонтане мутације.