Шта је Марфанов синдром?
Марфанов синдром описује комплексан насљедни поремећај везивног ткива, који углавном погађа очи, кардиоваскуларни систем и систем скелетних мишића. Међутим, имајући у виду да је сваки орган састављен од везивног ткива, Марфанов синдром може идеално уништити и значајно ометати раст и функцију сваког анатомског места.
Синдром се преноси као аутосомно доминантна особина: зато смо суочени са озбиљном генетском болешћу, која има изразито варијабилну фенотипску експресију (дефекти се могу веома разликовати од породице до породице или од пацијента до пацијента).
Оно што изазива Марфанов синдром је промена ФБН1 гена (на хромозому 15), који кодира фибрилин-1, веома важан везивни гликопротеин који чини структурну подршку за микрофибриле.
Микрофибриле: које се састоје од фибрилина, микрофибриле су присутне у екстрацелуларном матриксу, у коме формирају мрежу за таложење еластина у еластичним влакнима. Иако је свеприсутна у телу, микрофибриле обилују углавном у аорти, лигаментима и зонилима цилијарних тела (на нивоу ока).
Као аутосомно доминантна болест, само деца која су наследила ген за ФБН-1, промењена од оба родитеља, погођена су Марфановим синдромом. Ипак, у једном од четири случаја болест је резултат спонтаних мутација код пацијената који немају породичну историју.
Име болести долази од француског педијатра који га је први пут описао 1896. године (А. Марфан), након чега је морао да сачека до 1991. да идентификује измењени ген укључен у симптоматску манифестацију: откривач је био Ф. Рамирез.
Погледајте видео
к Погледајте видео на иоутубеuzroci
Споменули смо да је Марфанов синдром непосредан израз мутације гена који кодира фибрилин-1. Хајде да покушамо да продубимо тему да бисмо разумели који механизам је успостављен да покрене синдром.
ФИБРИЛЛИНА 1 је гликопротеинска компонента еластина, неопходна за обезбеђивање и одржавање еластичности и чврстоће ткива. У физиолошким условима, фибрилин 1 се везује за други протеин, познат као ТГФ-бета (или трансформирајући бета фактор раста). Чини се да је ТГФ-бета укључен у штетне процесе који утичу на васкуларни глатки мишић и екстрацелуларни матрикс. Полазећи од ових претпоставки, неки аутори су убеђени да је Марфанов синдром, поред мутације гена ФБН-1, последица и вишка ТГФ-бета, посебно у аорти, у срчаним залисцима и плућима.
учесталост
Процењује се да Марфанов синдром утиче на 1 субјекат на сваких 3.000-5.000 рођења и манифестује се нејасно између мушкараца и жена, без склоности ка прецизној раси. Статистике показују да 75% пацијената има позитивну породичну историју; у преосталих 25% узрок лежи у спорадичним мутацијама које се на неки начин повезују са старијим узрастом оца у време зачећа.
Деца са екстремно тешким облицима Марфановог синдрома имају очекивани животни век краћи од једне године.
Пре еволуције хируршких стратегија на отвореном срцу, већина пацијената са Марфановим синдромом имала је просечан животни век од 32 године; Захваљујући сталном побољшању медицинских и фармаколошких терапија, тренутно пацијенти са Марфановим синдромом живе у просеку до 60 година.
Знаци и симптоми
Да бисте сазнали више: Симптоми Марфанов синдром
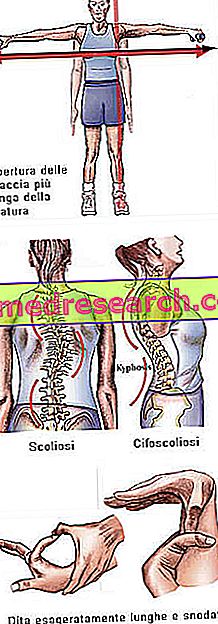
Марфанов синдром може да ради потпуно асимптоматски. Оболели пацијенти имају изразито витку структуру, несразмерно високу и танку. Доњи и горњи удови имају много већу дужину од дебла (доликостеномегалија). Такође се говори о аракодактији која најбоље изражава концепт претјеране дужине прстију, типичног за субјекте погођене Марфановим синдромом: дакле, руке су упоређене са ногама паука.
Што се тиче висине, ови пацијенти имају просечну висину изнад 97. перцентила.
Остале карактеристике које се често јављају код пацијената са Марфановим синдромом су:
- Руке отворене веће од висине
- Лоосе зглобови → претјерана покретљивост зглобова
- Деформација грудног зида
- Цристаллине дисплацемент
- Горњи део тела је мање развијен од доњег дела
- Спонтани пнеумоторакс (11%)
- сколиоза
- Стрије коже на бедру, леђима, делтоидном, пекторалном нивоу
Међу најпроблематичнијим знаковима повезаним са Марфановим синдромом, спомињемо пролапс срчаног залиска и инсуфицијенцију митралне валвуле: слично стање може лако да доведе до ширења аортног прстена и дисекције аорте.
Табела показује знакове који се могу наћи код пацијената са Марфановим синдромом. Описани ликови нису увек присутни, али се добар део њих може наћи.
| Аффецтед анатомиц сите | Могући симптоми |
сладак | Стрије у торакалном, лумбалном и сакралном подручју |
очи | Оштећење вида, астигматизам, одвајање ретине, уски кут глаукома, дислокација сочива, миопија |
Боне струцтуре | Артралгија, кифосколиоза, долицостеномелија (прекомерни развој дужине удова у односу на дебло), хипермобилност, високо непце, деформисани прсни кош, равне ноге, уски и танки зглобови, абнормални повратак / избочење на стернум, сколиоза, закривљена рамена, спондилолистеза |
дита | арацхнодацтили |
плућа | Спонтани пнеумоторакс, диспнеја, идиопатска плућна опструктивна болест |
Фациал цхангес | Огивално непце (малформација непца), мандибуларна ретрогнатија (дефект у развоју чељусти), издужено лице |
срце | Ангина пекторис, анеуризма абдоминалне аорте, срчана аритмија, дилатација / руптура / дисекција торакалне аорте, аортна инсуфицијенција, пролапс митралног залиска |
језик | Тежина језика |
дијагноза
Узимајући у обзир више од 200 могућих мутација, употреба генетских маркера је готово немогућа за дијагностичке сврхе.
Констатација Марфановог синдрома није увијек тако непосредна, с обзиром да фенотипска експресија мутације није увијек очигледна и једноставна за идентификацију. Дијагностичко кашњење може озбиљно угрозити опстанак пацијента: само помислите, на пример, на неуспех да се препозна кардиоваскуларни проблем.
Дијагностички критеријуми за Марфанов синдром израђени су на међународном нивоу 1996. године: дијагноза се састоји у истраживању породичне анамнезе повезане са комбинацијом главних и мањих индикатора синдрома.
Неки од многих дијагностичких тестова који се користе су:
- ehokardiogram
- Магнетна ангиорезонација и ЦТ (за испитивање аорте)
- магнетна резонантна ангиографија (МРА) са контрастном течношћу (како би унутрашње структуре аорте биле очигледне)
- испитивање са прорезаним лампама (за анализу могућег померања сочива)
- мерење очног притиска (да се истакне могуће присуство глаукома)
- генетски тестови (препоручује се пре заснивања детета да се утврди синдром или не)
терапије
Будући да је генетска болест, не постоји лек или третман који може преокренути болест.
Употреба лекова је, међутим, од суштинског значаја за ублажавање симптома и избегавање било каквих компликација, посебно срчаних. У ту сврху посебно су погодни лекови за смањење крвног притиска, као што су сартани (нарочито), АЦЕ инхибитори и бета-блокатори.
У контексту Марфановог синдрома, пацијенти који пате од сколиозе могу да прате специфичну негу, као и оне који су погођени глаукомом.
Хирургија је замислива да би се исправила аномална дилатација аорте, елемент који често уједињује већину пацијената са Марфановим синдромом.
Наставак: Марфанов синдром - лекови и нега »


